
HERVE AVET-LOISEAU & JILL CORRE
Altérations Moléculaires Et Hétérogénéité Au Sein Des Cellules Myélomateuses.
Notre objectif, principalement translationnel, est d’améliorer la prise en charge thérapeutique des patients atteints de myélome multiple (MM), un cancer de la moelle osseuse, et de prolonger leur survie. Pour ce faire, nous avons créé un réseau national qui nous a permis de constituer la plus grande bio-banque de tumeurs au monde, regroupant les caractéristiques cliniques et évolutives de milliers de patients.
Traitement personnalisé en fonction des modifications chromosomiques.
Ces dernières années, de nombreux nouveaux médicaments ont été enregistrés, rendant le choix thérapeutique complexe pour les médecins. Notre objectif est de les guider dans le choix de la bonne combinaison de médicaments pour les bons patients. Plus précisément, nous voulons améliorer l’identification des patients à haut risque en cette ère de nouveaux médicaments, car les stratégies les plus efficaces profiteront surtout aux patients à haut risque nouvellement diagnostiqués. Récemment, nous avons décrit un score basé sur 6 marqueurs pour stratifier de manière précise et routinière les patients atteints de MM (Perrot et al, JCO 2019). Nous séquençons massivement ces tumeurs afin d’identifier les sous-groupes de patients les plus susceptibles de répondre à des traitements spécifiques, afin d’aider les cliniciens dans leurs choix. Nous avons récemment montré que 60% des patients en rechute précoce ne présentent aucune altération génomique pronostique connue. Notre projet vise à identifier, par séquençage à haut débit, les nouvelles anomalies génomiques qui prédisent le résultat clinique des patients atteints de myélome.
Évolutions sous-clonales dans le myélome
L’identification des sous-clones du MM a considérablement changé notre vision des mécanismes qui sous-tendent la résistance du MM. Cependant, l’hétérogénéité clonale du MM reste mal définie, principalement en raison de limitations techniques. Nous avons récemment acquis une plateforme Chromium de 10xGenomics, permettant l’analyse transcriptomique, CNV, et ATACSeq au niveau de la cellule unique (sc). En utilisant cette approche « multi-omique », nos objectifs sont de décrire le paysage sous-clonal de chaque patient au moment du diagnostic et de leur première rechute, et d’analyser les cellules tumorales résiduelles au moment de la réponse au traitement afin de déterminer quels sous-clones ont été sélectionnés. En intégrant des données omiques unicellulaires variables (ARN, CNV, ATAC, mutations), nous serons en mesure de comprendre les déterminants moléculaires de cette sélection. Les données obtenues pourraient avoir un impact majeur sur notre compréhension de la sélection sous-clonale avec un traitement donné (sélection induite par le traitement ou histoire naturelle), et si les sous-clones sélectionnés précocement seront les principaux au moment de la rechute.
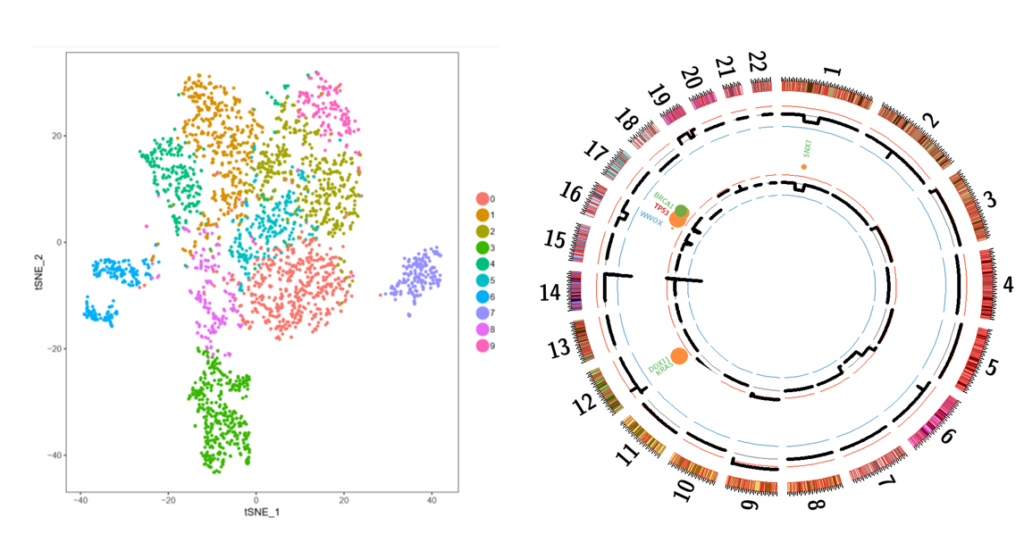
Figure 1 : Le séquençage d’ARN à cellule unique identifie différents sous-clones de MM au moment du diagnostic et de la rechute. Graphique t-SNE représentatif des cellules CD138+ de myelome au moment du diagnostic. Séquençage ciblé des cellules tumorales d’un patient au diagnostic (cercle extérieur) et à la rechute (cercle intérieur).

Figure 2 : Survie globale des patients en fonction du délai de la première rechute (tardive en rouge, précoce en bleu) et des facteurs de risques cytogénétiques connus (faible risque trait continu, haut risque trait discontinu).